فنیل کتونوری یک بیماری ژنتیکی و نقص متابولیکی مادرزادی نادر است که در آن سطوح اسید آمینه فنیل آلانین در مایعات و سیستم عصبی بدن بیمار تجمع مییابد. در واقع در افراد مبتلا به این بیماری یک آنزیم که مسئول تجزیه و تبدیل فنیل آلانین به تیروزین است وجود ندارد بنابراین وقتی فرد مواد خوراکی حاوی این اسید آمینه را که در غذاهای پروتئینی یافت میشود، مصرف میکند، به دلیل فقدان آنزیم تجزیه کننده، سطح بالایی از فنیل آلانین در بدن او تجمع مییابد. فنیل کتونوریا یک بیماری مادم العمر است و در صورت عدم درمان، فنیل کتونوری میتواند بر رشد شناختی فرد تأثیر بگذارد. درمان با داروها و یا تغییرات رژیم غذایی به کاهش علائم کمک میکند. اگر قصد دارید اطلاعات بیشتری در خصوص این بیماری ژنتیکی کسب کنید تا انتهای این مقاله علمی و مفید با تیم متخصص پینو بیبی همراه باشید.
بیماری فنیل کتونوری (Phenylketonuria) چیست؟
فنیل کتونوری (PKU) یک بیماری ژنتیکی است که باعث افزایش سطح مادهای به نام فنیل آلانین در بدن بیمار میشود. فنیل آلانین به عنوان بخشی از مسیرهای طبیعی بیوشیمیایی در بدن یافت میشود، اما مشکلات زمانی ایجاد میشوند که سطح آن به طور مداوم بالاتر از حد طبیعی باشد.
فنیل آلانین یک اسید آمینه است. آمینو اسیدها مولکولهایی هستند که پروتئینها را تشکیل میدهند. بسیاری از غذاهایی که میخوریم حاوی پروتئین و شیرین کننده مصنوعی آسپارتام هستند، که حاوی فنیل آلانین هستند. از این رو اگر این وضعیت درمان نشود، تجمع فنیل آلانین در بدن شما باعث ایجاد علائمی از جمله چالشهایی در رشد شناختی مانند ناتوانی ذهنی خواهد شد.

انواع فنیل کتونوری (PKU) چیست؟
بر اساس شدت تشخیص، انواع مختلفی از فنیل کتونوری (PKU) وجود دارد. علائم در میان موارد شدید در کسانی که درمان نشدهاند بدتر است. انواع PKU عبارت هستند از:
- PKU کلاسیک (شدیدترین حالت بیماری)
- PKU سطح متوسط یا خفیف بیماری
- هیپر فنیل آلانینمی خفیف (با حداقل شدت)
فنیل کتونوری (PKU) چه کسانی را تحت تاثیر قرار میدهد؟
فنیل کتونوری (PKU) میتواند هر فردی را که دارای جهش در هر دو نسخه از ژن PAH باشد، مبتلا کند. مطالعات نشان میدهد که خطر بالاتری در میان افراد بومی آمریکایی یا اروپایی وجود دارد. اگر یک فرد مبتلا به PKU کنترل نشده در دوران بارداری دارای سطح بالایی از فنیل آلانین باشد، این میتواند باعث ناتوانی ذهنی، نقایص مادرزادی و سایر مشکلات در نوزادش شود، حتی اگر مادر مبتلا به فنیل کتونوری نباشد کودکش مبتلا PKU خواهد شد.
فنیل کتونوری (PKU) چقدر شایع است؟
در ایران سالانه ۳۰۰ تا ۴۰۰ کودک مبتلا به فنیل کتونوری متولد میشوند یعنی حدود یک نوازد از هر ۵۰۰۰ تولد زنده. در ایالات متحده امریکا، فنیل کتونوری (PKU) هر سال از هر ۱۰۰۰۰ تا ۱۵۰۰۰ نوزاد یک نفر را مبتلا میکند.
علائم فنیل کتونوری (PKU) چیست؟
کودکان مبتلا به PKU درمان نشده در بدو تولد طبیعی به نظر میرسند. اما در سن ۳ تا ۶ ماهگی، آنها شروع به از دست دادن علاقه به اطراف خود میکنند. در سن یک سالگی، رشد کودکان به تاخیر میافتد و پوست آنها نسبت به افراد بدون این عارضه رنگدانه کمتری دارد. اگر افراد مبتلا به PKU، فنیل آلانین را در رژیم غذایی خود محدود نکنند، دچار ناتوانیهای ذهنی و رشدی شدید میشوند. از آنجایی که تشخیص و درمان فنیل کتونوری (PKU) اغلب اندکی پس از تولد به دلیل غربالگری غیرطبیعی نوزاد رخ میدهد، علائم قابل توجه بسیار نادر است. علائم در افرادی که موارد تشخیص داده نشده یا درمان نشده دارند، شدیدتر و حادتر است. علائم فنیل کتونوریا درمان نشده عبارت هستند از:
- بثورات پوستی، مانند اگزما
- تغییر رنگ پوست و یا مو (در مقایسه با سایر اعضای خانواده، پوست روشنتری دارند و عنبیه چشمان این افراد عموماً به دلیل فقدان رنگدانهها آبی و یا روشن است. این روشنی پوست و مو به دلیل ناتوانی بدن در تبدیل فنیل آلانین به ملانین، رنگدانه مسئول تولید رنگ در بدن فرد است).
- اندازه سر کوچک (میکروسفالی)
- بوی کپک زدگی در نفس، پوست یا ادرار این کودکان
- بوی شبیه لاک ناخن در ادرار
علائم شدید فنیل کتونوری درمان نشده عبارت هستند از:
- مشکلات رفتاری یا اجتماعی
- تاخیر در رشد (کاهش یا کندی رشد)
- ناتوانیهای ذهنی
- تشنج، لرزش یا حرکات تکان دهنده در بازوها و پاها (البته این علائم نادر است)
کودکان و بزرگسالان مبتلا به هیپر فنیل آلانینمی خفیف در صورت عدم درمان در معرض خطر بسیار کمتری برای ناتوانیهای ذهنی هستند.
چه چیزی باعث فنیل کتونوری (PKU) میشود؟
جهش در هر دو نسخه از ژن PAH باعث فنیل کتونوری (PKU) میشود. ژن PAH به بدن دستور میدهد تا آنزیمی به نام فنیل آلانین هیدروکسیلاز (PAHase) بسازد که مسئول تبدیل اسیدهای آمینه به اجزای (پروتئین) است که بدن انسان میتواند از آنها استفاده کند. وقتی بدن انسان نمیتواند آمینو اسیدهایی را که در رژیم غذایی میخورد پردازش کند، در خون و بافتهای بدن تجمع مییابند. بدن انسان به فنیل آلانین حساس میشود. تجمع بیش از حد فنیل آلانین در بدن انسان به مغز او آسیب میرساند.
آیا ژن فنیل کتونوری (PKU) غالب است یا مغلوب؟
فنیل کتونوری (PKU) یک بیماری ژنتیکی است که والدین این ژنها را در الگوی اتوزومال مغلوب به کودکان منتقل میکنند. این بدان معنی است که نوزادان یک کپی از ژن جهش یافتهای را که باعث ایجاد PKU میشود از هر یک از والدین در زمان لقاح دریافت میکنند. در بیشتر موارد، والدین ناقل این ژن هستند اما علائم این بیماری فنیل کتونوری را ندارند.
در مقاله مربوط به ارث رسیدن صفات از والدین به کودک توضیحاتی درباره نحوه وراثت ژنها ارائه شد اما شاید توضیح برخی اصطلاحات در اینجا به درک بهتر نحوه انتقال ژن از والدین به نوزاد کمک کند. اتوزومال یعنی ژنهای مربوط به یک صفت روی کروموزومهای غیرجنسی قرار دارند و ربطی به جنسیت افراد ندارد. ژنها وقتی در بدن بیان میشوند یعنی آن صفت را نشان میدهند، رفتار ژنها نسبت به همدیگر حالت غالب و مغلوبی دارد. برای بروز یک صفت به حالت غالب یک ژن از سمت یکی از والدین کافی است و برای بروز ژن مغلوب باید کودک از هر دو والد ژن مغلوب را دریافت کند. اگر فقط یک کپی از ژن معیوب (مغلوب) در بدن والدین باشد ناقل آن بیماری هستند ولی الزاماً آن بیماری را نشان نمیدهند. البته استثناهایی در این میان وجود دارد که خارج از بحث این مقاله است.
چگونه فنیل کتونوری (PKU) تشخیص داده میشود؟
پزشک تشخیص فنیل کتونوری (PKU) را کمی پس از تولد به عنوان بخشی از غربالگری معمول نوزاد از طریق آزمایش خون تأیید میکنند. اگر سطح فنیل آلانین نوزاد در نمونه خون او بالا باشد، پزشک آزمایشهای بیشتری را برای تأیید تشخیص و نوع PKU انجام میدهد، معمولاً با آزمایشهای اضافی خون یا ادرار، این تشخیص کامل میشود. از آنجایی که فنیل کتونوری یک بیماری ژنتیکی است، آزمایش ژنتیکی میتواند جهش مسئول علائم را مشخص کند. اگر چه اکثر تشخیصهای PKU اندکی پس از تولد نوزاد اتفاق میافتد، اما اگر غربالگری نوزادان کامل نشده باشد، پزشکان میتوانند فنیل کتونوری را در هر سنی تشخیص دهند.
آزمایش ژنتیک برای تشخیص PKU
میتوان از نمونه خون برای آزمایش جهشهایی که باعث فنیل کتونوری PKU میشوند استفاده کرد. آزمایشات در سه سطح قابل انجام است:
- آزمایش نوزاد: آزمایش خونی که فنیل آلانین موجود در خون نوزاد را اندازهگیری میکند برای کمک به تشخیص PKU کافی است. بنابراین آزمایش DNA ضروری نیست. با این حال، اگر آزمایش PKU کودک مثبت باشد، پزشکان ممکن است آزمایش ژنتیک را توصیه کنند زیرا شناسایی نوع جهش درگیر میتواند به انتخاب مناسبترین طرح درمانی کمک کند. همچنین در صورتی که والدین هر دو ناقل PKU هستند و آزمایش خون استاندارد نوزاد این بیماری را نشان نمیدهد، باید آزمایش DNA روی کودک انجام شود. در صورتی که جهشهای عامل بیماری در خانواده شناسایی شده باشد، این آزمایش به طور قطعی فنیل کتونوری PKU را نشان میدهد یا رد میکند.
- آزمایش در دوران بارداری: یک زن باردار میتواند آزمایش DNA قبل از تولد را درخواست کند تا بفهمد آیا فرزندش با PKU متولد میشود یا خیر؟! برای انجام این آزمایش، یک پزشک برخی از سلولها را از طریق آمینوسنتز یا پرزهای کوریونی جهت آزمایش استخراج میکند. یک مشاور ژنتیک میتواند خطرات و مزایای آزمایش ژنتیکی را برای افراد تشریح کند و میتواند به توضیح گزینههای موجود برای آزمایش کمک کند. این بحث ممکن است به ویژه برای والدینی که یک فرزند مبتلا به PKU دارند مفید باشد، زیرا شانس ابتلا شدن فرزند بعدی آنها بالاتر از حد متوسط است.
- آزمایش حاملهای احتمالی PKU: اگر کودکی مبتلا به PKU تشخیص داده شود، سایر اعضای خانواده ممکن است احتمال بیشتری برای داشتن بچههایی مبتلا به PKU در آینده باشند. آنها باید آزمایش DNA نیز انجام دهند.
آیا آزمایشهای غربالگری برای تشخیص فنیل کتونوری (PKU) وجود دارد؟
آزمایش غربالگری فنیل آلانین، سطح فنیل آلانین را در خون مشخص میکند. نوزادان این آزمایش را بین ۲۴ تا ۷۲ ساعت پس از تولد به عنوان بخشی از گزارش سلامت نوزاد دریافت میکنند. پزشک با زدن یک سوزن کوچک از پاشنه پای کودک نمونه خون میگیرد. فقط چند قطره خون برای این آزمایش لازم است.
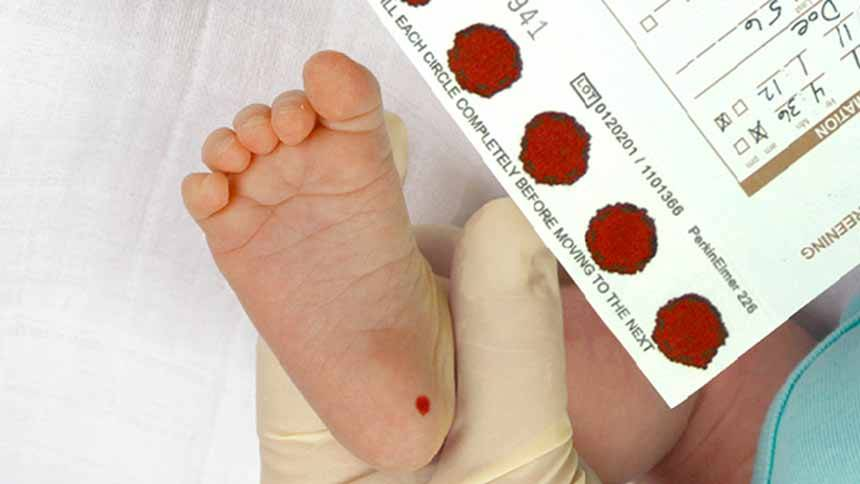
شرایط لازم برای دریافت نمونه جهت آزمایش فنیل کتونوری
برای تشخیص فنیل کتونوری در بزرگسالان و کودکان نمونههایی از خون و بافت گرفته میشود. نمونه مایع آمنیوتیک و پرزهای کوریونی برای دوران جنینی و بررسی احتمال ابتلا جنین به فنیل کتونوری در دوران جنین و بدو تولد است.
نمونه خون
- نیازی به ناشتا بودن قبل از آزمایش نیست.
- عدم استفاده از داروهای هورمونی و آنتی بیوتیکها حداقل سه روز قبل از روز آزمایش ضروری است.
- عدم مصرف داروهای اعصاب (در صورت مصرف این داروها احتمال تکرار آزمایش را بالا میرود)
- در بیماران هموفیلی و نقص فاکتور انعقادی خون باید قبل از آزمایش، فاکتور انعقادی تزریق شود.
- اگر بیمار سابقه پیوند مغز استخوان در ۶ ماه یا یک سال گذشته را داشته است باید به آزمایشگاه اطلاع بدهد.
نمونه بافت
قبل از نمونه برادری بافتی (بیوپسی) ممکن است مصرف برخی داروها نظیر آسپرین، داروهای ضدالتهابی غیر استروئیدی متوقف شود.
نمونه مایع آمنیوتیک (آمینوسنتز)
- نوشیدن آب فراوان قبل از نمونهگیری برای پیشگیری از کاهش حجم مایع آمنیوتیک اطراف جنین الزامی است.
- این آزمایش بین هفته ۱۵ تا ۲۰ بارداری انجام میشود.
نمونه پرز کوریونی CVS
- جهت نمونهگیری از پرز کوریونی CVS لوله نمونهگیری را معمولاً از طریق واژن وارد کرده، از پرزهای پرده کوریون نمونهگیری میشود.
- این آزمایش بین هفته ۱۰ تا ۱۳ بارداری انجام میشود.
آیا کودک مبتلا به PKU نیاز به آزمایش مکرر دارد؟
نوزادان و کودکان مبتلا به PKU به انجام متناوب آزمایش خون جهت اندازهگیری فنیل آلانین در خون خود نیاز دارند. اگر شواهدی از فنیل آلانین بیش از حد یا خیلی کم وجود داشته باشد، ممکن است پزشک تغییراتی را در رژیم غذایی یا شیر خشکی که کودک دریافت میکند پیشنهاد دهد.
نوزادان مبتلا به PKU حدود یک بار در هفته در سال اول زندگی خود آزمایش خواهند شد. پس از سال اول، کودکان ممکن است یک یا دو بار در ماه آزمایش شوند. بزرگسالان نیز باید در طول زندگی خود به طور منظم معاینه شوند. اغلب، نمونه خون را میتوان در خانه گرفته و به آزمایشگاه ارسال کرد.
فنیل کتونوری (PKU) چگونه درمان میشود؟
درمان فنیل کتونوری؛ یک درمان مورد نیاز فرد مبتلا به صورت مادام العمر است. درمان قطعی برای فنیل کتونوری و اصولاً بیماریهای ژنتیکی وجود ندارد بلکه راهکارهای درمانی در طول مسیر زندگی فرد، علائم بیماری را بهبود میبخشند. درمان در فنیل کتونوری ممکن است شامل یک رژیم غذایی خاص یا دارو باشد.
درمان میتواند شامل موارد زیر باشد:
- خوردن یک رژیم غذایی خاص کم فنیل آلانین اما سرشار از مواد مغذی.
- مصرف ویتامینها، مواد معدنی و مکملها.
- افزودن یک داروی مکمل به نام ساپروپترین دی هیدروکلراید (Kuvan®) برای تجزیه فنیل آلانین در بدن بیمار.
- دارویی به نام Pegvaliase (Palynziq®) به افراد مبتلا به PKU این امکان را میدهد که رژیم غذایی نامحدودی را بدون هیچ مکمل یا کووان مصرف کنند. این دارو جایگزین آنزیمی میشود که به تجزیه فنیل آلانین کمک میکند، که در مبتلایان به فنیل کتونوری (پی کا یو) به درستی کار نمیکند.

چه غذاهایی حاوی فنیل آلانین هستند؟
فردی که مبتلا به فنیل کتونوری (PKU) تشخیص داده شده است، نیاز به خوردن نوع خاصی از غذاها را دارد. رژیم غذایی برای محدود کردن مقدار غذاهای حاوی فنیل آلانین است (اگر فنیل کتونوری آنها با پگوالیاز درمان نمیشود). فنیل آلانین را در غذاهای سرشار از پروتئین، از جمله موارد ذیل یافت میشود:
- محصولات گندم مثل ماکارانی و نودل
- لبنیاتی مانند شیر، پنیر
- تخم مرغ
- آجیل و خشکبار
- کره بادام زمینی
- گوشتها مانند ماهی، جوجه، گوشت گاو
- لوبیا، نخود فرنگی
- ژلاتینها و پاستیل
- شیرین کننده مصنوعی (آسپارتام) در نوشابهها و آدامس یافت میشوند.
آیا رژیم غذایی برای فنیل کتونوری (PKU) وجود دارد؟
اگر فنیل کتونوری با پگوالیاز درمان نشوند، یک رژیم غذایی کم پروتئین برای افراد مبتلا به فنیل کتونوری (PKU) بهترین راهکار درمانی است. فرد مبتلا باید از غذاهای حاوی پروتئین مانند گوشت، تخم مرغ و محصولات لبنی، حتی گیاهانی که میزان پروتیئن بالایی دارند مثل لوبیا و سویا پرهیز کند. اگر کودک یا والدین مبتلا به PKU هستند، باید با پزشک یا یک متخصص تغذیه در مورد تنظیم یک رژیم غذایی متعادل و پر از مواد مغذی ضروری مشورت شود تا علاوه بر یک زندگی سالم، زندگی لذت بخش نیز داشته باشند.
چگونه میتوان از فنیل کتونوری (PKU) جلوگیری کرد؟
هیچ راهی برای پیشگیری از فنیل کتونوری (PKU) وجود ندارد. اگر قصد باردار شدن دارید و میخواهید خطر داشتن فرزندی با یک بیماری ژنتیکی را متوجه شوید، با پزشک خود در مورد آزمایش ژنتیک مشورت کنید.
چشمانداز فرد مبتلا به فنیل کتونوری (PKU)
فنیل کتونوری (PKU) یک بیماری مادام العمر است و اکثر افرادی که با آن زندگی میکنند زندگی سالمی دارند. پس از تشخیص، آزمایش خون منظم برای نظارت بر سطح فنیل آلانین در خون فرد ضروری است. نیاز است تا بسیاری از افراد مبتلا به این بیماری از مشاوره و راهنماییهایی یک متخصص تغذیه بهره ببرند؛ متخصص تغذیه میتواند در مورد این که بهترین غذاها برای این افراد چیست و از چه مواد غذایی باید اجتناب کنند؛ آنها را راهنمایی کند تا مطمئن شوند که تمام مواد مغذی مورد نیاز برای سالم ماندن را دریافت خواهند کرد.
اگر فردی مبتلا به PKU و باردار میباشد، پزشک و یا متخصص تغذیه یک برنامه غذایی خاص برای او تنظیم میکنند. تغذیه مناسب سلامتی فرد را تضمین میکند و در عین حال خطر عوارض بعدی در نوزاد را نیز کاهش میدهد.
خودمراقبتی در افراد مبتلا به فنیل کتونوری
تشخیص فنیل کتونوری (PKU) مادام العمر است، بنابراین لازم است تا فرد مبتلا همواره با پزشک همراه خود در تماس باشد و آزمایشات خون معمولی را برای نظارت بر سطح فنیل آلانین در بدن انجام دهد.
از آنجا که رژیم غذایی این افراد محدود است، برای جبران کمبود پروتئین در رژیم غذایی باید جیره غذایی مناسبی داشته باشند و ویتامینهای مورد نیاز بدنشان را مصرف کنند. فرد مبتلا باید رژیم غذایی را برای تمام عمر خود رعایت کند تا از علائمی که میتواند برای سلامتی او تاثیر بگذارند و خطرناک باشد جلوگیری کند.
زمان مراجعه به پزشک برای فنیل کتونوری
اگر والدین یا فرزندشان علائم فنیل کتونوری را دارند، برای ارزیابی کامل با پزشک خود تماس بگیرند. قبل از باردار شدن یا در طول اولین معاینه قبل از تولد، میتوانند از پزشک خود در مورد آزمایش (که غربالگری حامل نامیده میشود) بپرسند تا تعیین کند آیا او و همسرش در معرض خطر داشتن فرزند مبتلا به PKU هستند یا خیر. اگر فردی مبتلا PKU است، برای بررسی وضعیت خود به پزشک مراجعه کند تا آزمایشهای خونی منظم انجام دهد.
چه سوالاتی باید از پزشک در مورد فنیل کتونوری بپرسید؟
- چگونه مطمئن شویم که کودکمان مواد مغذی کافی برای سالم ماندن و رشد دریافت میکند؟
- آیا نیاز به مصرف ویتامین یا مکمل است؟
- آیا یک رژیم غذایی کم پروتئین برای کاهش سطح فنیل آلانین کافی است یا نیاز به دارو نیز وجود دارد؟
سخن آخر
در مراحل اولیه تشخیص، ممکن است انتخاب غذاهایی که حاوی فنیل آلانین نیستند دشوار باشد. با پزشک و یا یک متخصص تغذیه همکاری نزدیکی داشته باشید تا به شما در ایجاد یک رژیم غذایی پر از مواد مغذی ضروری و در عین حال حذف فنیل آلانین کمک کند. PKU یک بیماری مادام العمر است و رعایت رژیم غذایی و منظم بودن قرار ویزیتهای با پزشک برای بررسی سطح فنیل آلانین در خون باعث ایجاد عادات مثبت و مادام العمر میشود که بر کیفیت زندگی فرد مبتلا بسیار موثر است.